Contents |
|---|
Markov Model |
Figures |
| Forensic mathematics web site |
Jérôme Buard, Institut de Génétique Humaine, Montpellier, France
Jerome.Buard@igh.cnrs.fr
Charles Brenner, Department of Public Health, Berkeley, USA
cbrenner@uclink.berkeley.edu
Sir Alec Jeffreys, Department of Genetics, Leicester, UK
ajj@le.ac.uk
Our model consists of a transition matrix T = (tsj), where tsj is the probability that the offspring of an allele of s repeats will have j repeats.
The model is constructed in these stages:
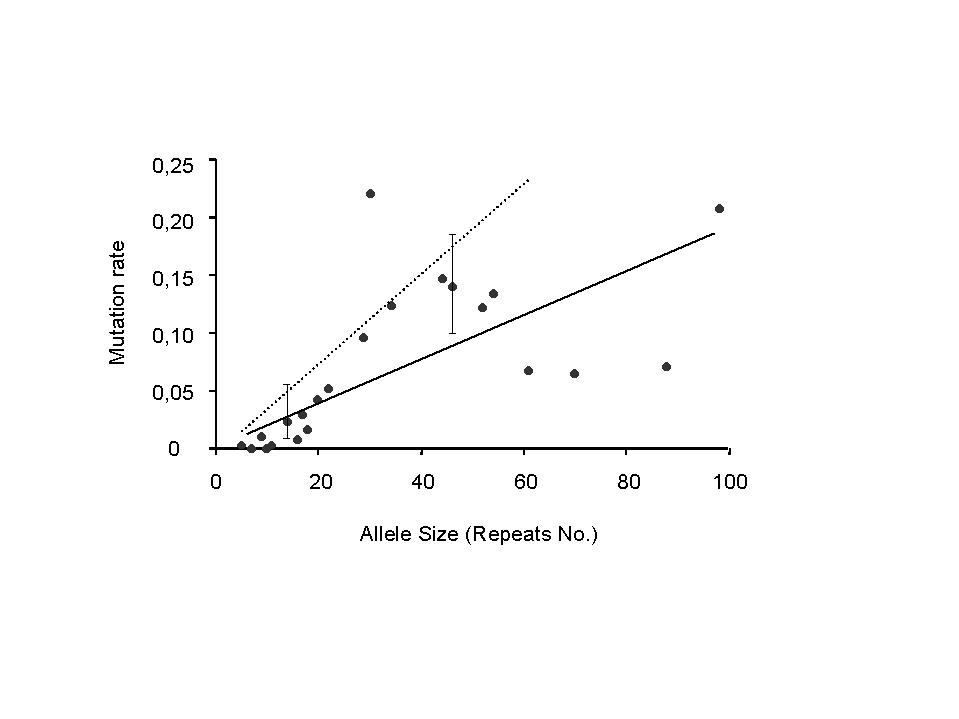
1.a m(s)=probability of mutation as a function of progenitor size s (figure 2a)
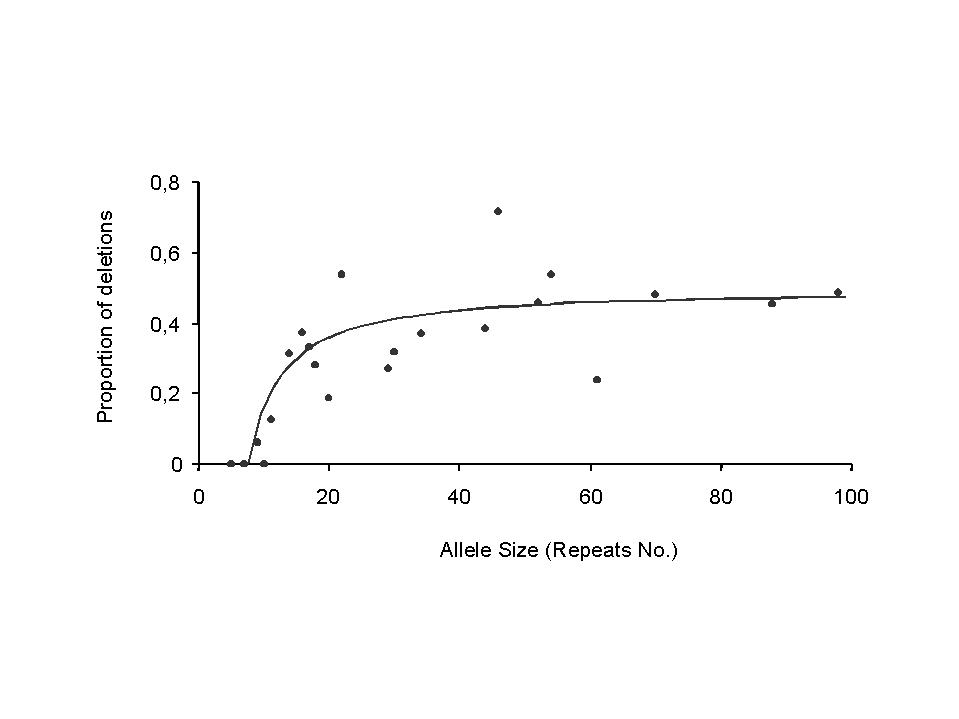
1.b d(s)=proportion of mutations that are contractions (figure 2b)
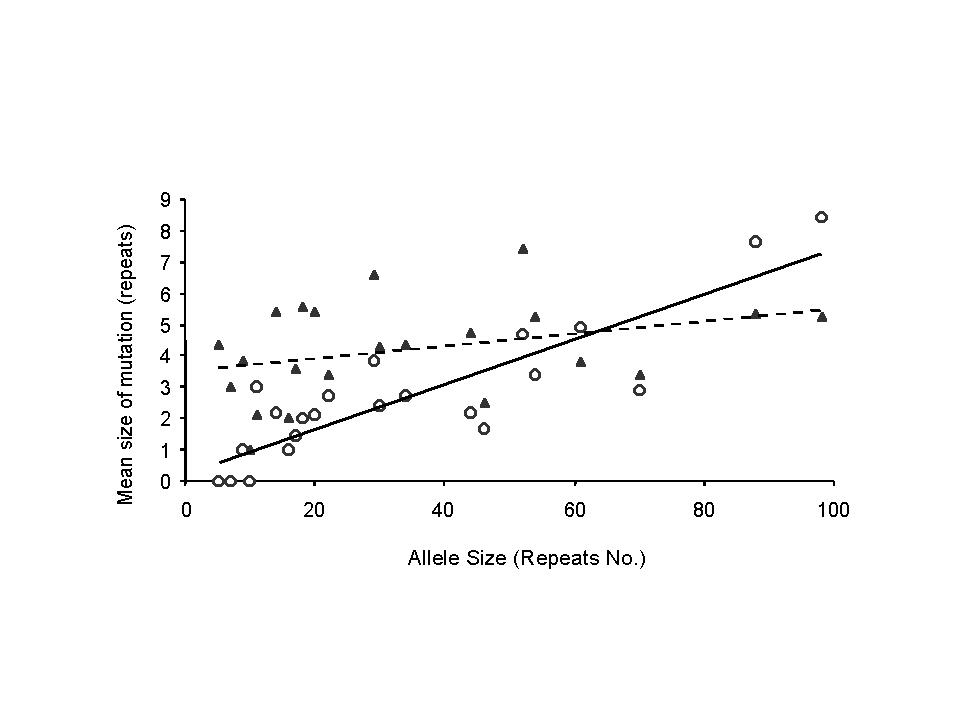
1.c D(s)=average contraction amount, assuming contraction occurs (figure 2c)
1.d E(s)=expansion amount, assuming expansion occurs (figure 2c)
1.e S(s, a)=probability to contract by a repeat units, assuming contraction occurs
1.f L(s, a)=probability to expand by a repeat units, assuming expansion occurs
Before giving details of the choices for each of the above functions, we discuss how T is built from them.
Denote by T=(tsj) a Markov matrix in which tsj is the transition probability for an allele of s repeats to mutate to j repeats in one generation. The transition of allele frequencies from one generation to the next is therefore given by a(g+1)=a(g)T.
The curves described in figure 2 determine the entries tsj.
From figure 2a, tss = 1–m(s). Since d(s) (see figure 2b) is the probability of deletions assuming mutation, the unconditional probability of deletion is m(s) d(s), which is SUMj<stsj. As for the distribution of these deletions as a function of deletion amount, we assume the slower-than-exponential decline that tsj proportional to exp[–h(s)(a–1)0.59] where a=|s–j| is the amount of deletion. Here h(s), h(s)>0, is determined by the condition that the expected deletion amount assuming deletion is D(s) (see figure 2c), i.e. that SUMj<s ats, s-a /[m(s)d(s)] = D(s). Similarly, E(s) and the model assumption that ts, s+a declines exponentially with a–1 determines tsj for j>s.
In summary, for j<s,
tsj=m(s)d(s)b(s)exp[–h(s)(a–1)0.59], b(s) being the normalizing constant 1/SUMj<s exp[–h(s)(a–1)0.59],
and for j>s, a function k(s) analogous to h(s) is determined such that
tsj=m(s)(1–d(s))c(s)exp[–k(s)(a–1)], where c(s) = 1/SUMj>s exp[–k(s)(a–1)].
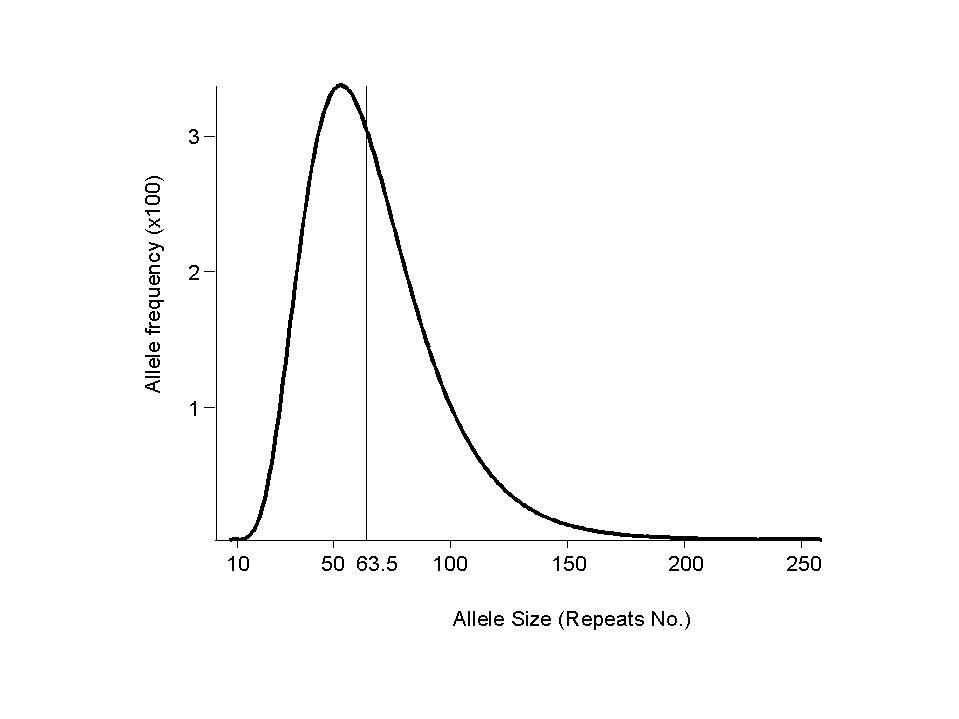
We begin with an arbitrary allele frequency distribution a(0) such as a40(0) = 1, as(0) = 0 for s not= 40. The allele distributions at successive generations are given by a(g)=a(0)tg. An equilibrium distribution a (such that a=aT), independent of a(0), is effectively attained within 1000 generations.
From the data of observed mutation rates for 23 progenitor alleles of various sizes, we developed a model in stages.
4.a mutation probability m(s) as a function of progenitor size s (figure 2a)
A linear regression, m(s)=as+b, gives b=1/75, but there is no evidence (p=0.45) from the data that b not= 0. If b=0, then the linear fit becomes m(s)=s/521, with considerable random variation (R2=0.42). A quadratic fit m(s)=s/303 – s2/50000 explains the data only a little better (R2=0.52) at the expense of an extra parameter. It is illogical anyway for it says that the mutation rate eventually declines (above 82 repeats) and even becomes negative (above 165 repeats). Therefore we choose the simple linear model m(s)=s/521.
4.b proportion d(s) of mutations that are contractions (expansions) (figure 2b)
Contractions and expansions approach equal frequency for large sizes, suggesting a model d(s)=0.5–k/s for contractions (and e(s)=0.5+k/s for expansions). A linear regression d(s)–0.5 as a function of 1/s gives a constant term of only 1/130, and the p-value of 0.8 as well as the attraction of symmetry further suggest constraining the extra constant to be 0. Therefore we obtain d(s)=0.5–3.28/s, R2=0.6. For s<7, we set d(s)=0.
4.c contraction amount, assuming contraction occurs, D(s) (figure 2c)
A linear regression gives D(s)=0.67 + s/15.4, for which R2=0.71. However, the proposition that the constant term is non-zero is only weakly supported by the data (p=0.17), and D(s)=s/13 explains the data nearly as well (R2=0.67). Therefore we choose the simpler formula.
4.d expansion amount, assuming expantion occurs, E(s) (figure 2c)
We choose to model E(s) by the linear regression E(s)=3.54 + s/50. It may be (p=0.12) that the linear term is unnecessary; the constant fit E(s)=4.2 fits the data only a little less well.
4.e distribution of contraction amounts
From the data, the mutation amount
a=|s–j| tends to be small. This suggests a model of the form
P(a repeat-unit contraction) proportional to
k(s)a–1, 0<k<1. However such a
model doesn't account for the occasional contraction by a large amount; therefore we chose a more
elaborate model wherein decay of mutation probabilities with a–1 is
slower-than-exponential. One way to accomplish this is to replace a–1 by
(a–1)beta for some beta<1.
For any given beta, for each s the value of k(s) is constrained by
the condition that the mean contraction amount must be D(s), and thus an expected
number of contractions S(s, a) for each s and each a
can be computed. The best-fit estimate for was obtained as that that gives the best least-squares
fit of S(s, a). The value obtained is beta=0.59.
4.f distribution of expansion amounts
For expansions, any value of beta in the range 0.9<beta<1.2 gives, within 2-decimal
accuracy, an equally good fit to the observed expansion amounts. Therefore, by Occam's Razor and
since beta>1 seems biologically implausible (suggesting some sort of anti-mutational spring that
makes a third step of mutation, given that a second has occurred, less likely than the chance to
mutate a second step, given that a mutation has occurred), we omit the extra parameter and assume
exponential decline.
In this section we discuss the relationship between various model assumptions and the
equilibrium distribution.
5.a mutation probability m(s) as a
function of progenitor size sSection 5: Robustness of the model
The equilibrium distribution is not sensitive to the choice of function m(s). Multiplying m(s) by a constant affects the time to equilibrium, but predictably has no perceptible affect on the distribution at all. Once this point is appreciated it is also not surprising that altering the shape of m(s) in other ways – for example capping m(s) as m(s)=min (s/521, 0.2) – also has little or no effect on the equilibrium distribution.
5.b proportion d(s) of mutations that are contractions (expansions)
Although the process described above for selecting d(s) seems compelling, to test the sensitivity of the model to d(s) we tried the effect of the model d(s)=0.47–constant/s. The effect is to decrease (!) slightly the mean allele size, and significantly increase the proportion of small (s<20) alleles.
5.c average amount D(s) of contraction, assuming contraction occurs (figure 2c)
The alternate model D(s)=0.67 + s/15.4 slightly increases the means size, compared to our selected model.
5.d expansion amount, assuming expantion occurs, E(s)
The alternate model E(s)=4.2 modestly decreases the means size, compared to our selected model.
5.e distribution of contraction amounts
Omitting beta from the model (i.e. choosing beta=1), slightly increases the mean allele size, and considerably reduces the incidence of small alleles. Either beta<1, or a complicated model of average mutation rate, seems necessary in order to explain the plethora of small alleles observed in collected population samples.
5.f distribution of expansion amounts
For symmetry we consider introducing the parameter beta=0.59 for expansions, as it is for contractions. This change would seem to encourage large expansion events, but paradoxically it modestly decreases the mean allele size at equilibrium, and significantly increases the number of small alleles.
| mean | st dev | skew | kurtosis | peak | mode | <10 | <20 | |||
|---|---|---|---|---|---|---|---|---|---|---|
| repeats | ||||||||||
| Simulation | ||||||||||
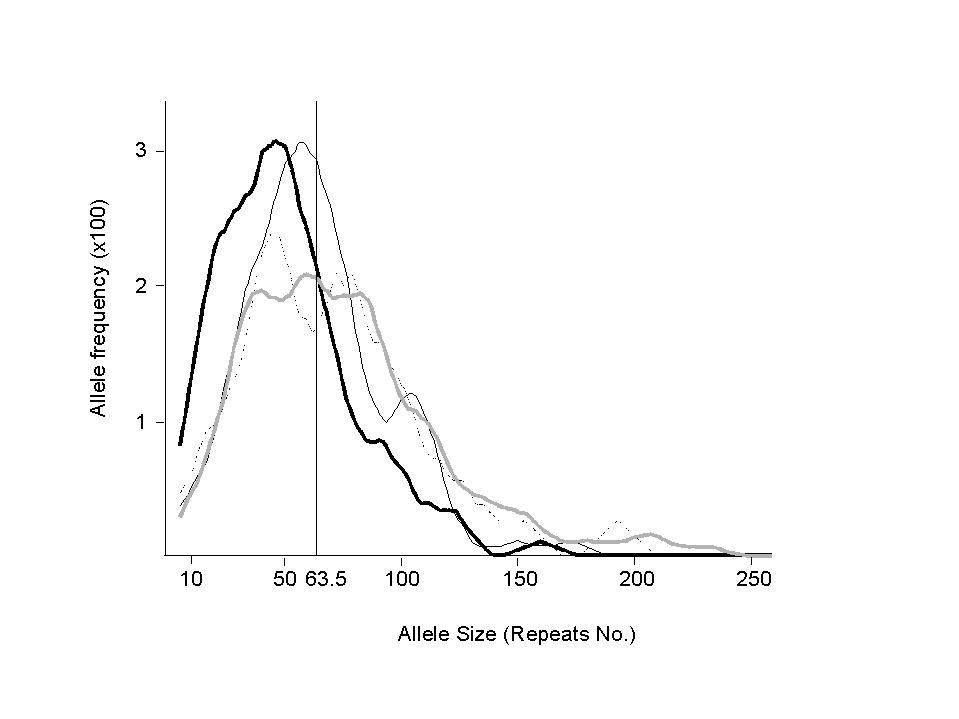
| standard model | 63.5 | 32 | 1.15 | 5.4 | 49 | 58 | 0.8% | 3.4% | ||
| double m(s) (to 2s/521) | no change | |||||||||
| shrink m(s) | 61.5 | 32.8 | 0.98 | 5.1 | 49 | 57 | 3.4% | 6.8% | ||
| beta=1 | 67.3 | 27.9 | 1.19 | 5.5 | 54 | 62 | 0.3% | |||
| both of preceding | 67.3 | 27.9 | 1.19 | 5.5 | 54 | 62 | 0.01% | 0.3% | ||
| beta=0.59 for expansions also | 59.7 | 34 | 1.18 | 5.8 | 45 | 54 | 3.7% | 7.7% | ||
| E(s)=constant | 57 | 24 | 0.6 | 3.7 | ||||||
| d(s)=0.47–cons/s | 60 | 32.3 | 0.99 | 5.1 | 47 | 56 | 3.5% | 7.2% | ||
| D(s)=0.67–s/15.4 | 66.3 | 35 | 0.17 | 5.4 | ||||||
| Populations | ||||||||||
| African | 51.4 | 28.7 | 0.93 | 4.1 | 51 | 48 | 2.5% | 10% | ||
| European | 78 | 47.3 | 1.78 | 8.6 | 93 | 70 | 1.2% | 4% | ||
| Indian | 80.4 | 59.7 | 2.23 | 8.9 | 47 | 72 | 2.1% | 6.3 | ||
| Japanese | 63.7 | 29.6 | 0.7 | 3.9 | 34 | 60 | 1.8% | 4.5% | ||
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}